Paparan sinar UV tinggi pada sel dapat mengakibatkan mutasi. Kalimat yang terdengar seperti teori pelajaran biologi itu ternyata adalah drama molekuler berkecepatan tinggi yang terjadi setiap kali kulit kita bertemu matahari. Bayangkan, foton-foton kecil dari sinar ultraviolet itu bukan sekadar memberi warna coklat, tapi menyelinap masuk hingga ke inti sel, tempat peta kehidupan kita—DNA—disimpan. Di sana, mereka mulai membuat kekacauan dengan cara yang licik dan terselubung, menyatukan huruf-huruf genetik yang tidak seharusnya bersatu.
Proses ini dimulai ketika energi dari sinar UV-B dan UV-A diserap oleh basa nitrogen dalam untai DNA. Penyerapan energi ini memicu reaksi kimia yang menyebabkan terbentuknya “dimer”, yaitu dua basa pirimidin yang berdekatan—seperti thymine—menyatu menjadi satu gumpalan yang abnormal. Struktur baru ini mengubah bentuk heliks DNA, bagai sebuah jalan raya yang tiba-tata memiliki polisi tidur yang besar dan tidak semestinya.
Kekacauan inilah yang menjadi awal dari rentetan kesalahan yang, jika tidak diperbaiki, akan terkunci menjadi mutasi permanen dalam kode genetik sel.
Mekanisme Terselubung Radiasi Ultraviolet dalam Mengacaukan Peta Genetika Sel
Ketika kita membicarakan sinar ultraviolet (UV) dari matahari, seringkali yang terbayang adalah kulit terbakar atau kemerahan. Namun, di balik efek kasat mata itu, terjadi sebuah drama molekuler yang sangat rumit di dalam inti setiap sel kulit kita. Drama ini bermula dari interaksi langsung antara foton berenergi tinggi dengan molekul paling berharga kita: DNA. Proses ini bukan sekadar pukulan kasar; ini adalah gangguan presisi tinggi yang mengubah cetak biru kehidupan sel.
Proses Fisikokimia Penyerapan Foton dan Pembentukan Dimer
Mekanisme ini dimulai ketika foton dari spektrum UV-B (280-315 nm) dan UV-A (315-400 nm) menembus lapisan kulit dan diserap oleh basa nitrogen dalam untai DNA. UV-B, dengan energinya yang lebih tinggi, merupakan pemain utama dalam kerusakan langsung. Foton UV-B memiliki energi yang tepat untuk dieksitasi oleh cincin pirimidin—yaitu sitosin dan timin—dalam DNA. Penyerapan energi ini menyebabkan elektron dalam basa tersebut tereksitasi ke keadaan yang lebih reaktif.
Dalam keadaan ini, ikatan karbon-karbon rangkap pada cincin pirimidin menjadi lebih rentan. Ketika dua pirimidin, khususnya dua timin, terletak berdekatan dalam urutan yang sama pada satu untai DNA, keadaan tereksitasi ini memungkinkan terbentuknya ikatan kovalen yang tidak normal di antara mereka.
Ikatan baru ini mengubah struktur lokal DNA secara dramatis. Dua basa yang seharusnya terpisah kini menyatu, membentuk struktur kaku yang disebut dimer pirimidin. Dimer timin adalah yang paling umum. Struktur bengkok ini menyebabkan distorsi heliks ganda yang signifikan; heliks DNA menjadi terpuntir dan tertekuk di lokasi kerusakan. Distorsi ini bukan hanya cacat struktural, tetapi juga merupakan penghalang fisik bagi mesin molekuler sel yang harus membaca dan mereplikasi informasi genetik.
Sementara UV-A, meski kurang energik, juga berkontribusi melalui mekanisme tidak langsung dengan menghasilkan spesies oksigen reaktif yang dapat merusak DNA dan mengganggu proses perbaikan, memperburuk efek dari UV-B.
| Jenis Kerusakan | Panjang Gelombang Pemicu Utama | Tingkat Mutagenesis | Efisiensi Perbaikan oleh Sel (NER) |
|---|---|---|---|
| Cyclobutane Pyrimidine Dimer (CPD) | UV-B (<320 nm) | Sangat Tinggi | Cukup Efisien, tetapi jumlah masif dapat membanjiri sistem. |
| Pyrimidine (6-4) Pyrimidone Photoproduct (6-4PP) | UV-B (<320 nm) | Tinggi | Lebih Efisien, karena distorsi heliks yang ditimbulkan lebih besar dan lebih mudah dideteksi. |
| Photooxidative Damage (8-oxoGuanine, dll.) | UV-A (dominan) | Sedang hingga Tinggi | Bervariasi, bergantung pada sistem perbaikan eksisi basa; bisa lolos jika tidak diperbaiki. |
Bayangkan DNA sebagai sebuah halaman teks yang sangat panjang, berisi instruksi untuk membangun dan menjalankan sel. Proses replikasi DNA ibarat mesin fotokopi yang sangat canggih yang membaca dan menduplikasi halaman tersebut. Dimer timin adalah seperti dua huruf “T” yang saling menempel dan membentuk satu gumpalan tinta yang besar dan kabur. Ketika mesin fotokopi (enzim DNA polimerase) sampai ke titik itu, ia tidak bisa membaca huruf individual mana yang seharusnya ada. Bingung, mesin itu mungkin akan memasukkan huruf yang salah di seberang gumpalan itu pada cetakan baru, atau justru macet dan berhenti bekerja. Kesalahan cetak inilah yang nantinya menjadi mutasi tetap dalam salinan genetik sel anak.
Proses Penguncian Mutasi Titik Setelah Gagal Perbaikan
Jika dimer pirimidin tidak diperbaiki sebelum sel memasuki fase sintesis DNA (S-phase), konsekuensinya menjadi permanen. Sel yang gagal memperbaiki kerusakan tersebut akan mengunci mutasi selama pembelahan sel berikutnya melalui serangkaian tahapan yang berurutan.
- Pertama, enzim DNA polimerase yang bertugas mensintesis untai baru selama replikasi menemukan dimer timin di untai templat. Dimer ini menghalangi polimerase untuk melanjutkan sintesis secara normal karena struktur fisiknya yang menyimpang.
- Kedua, untuk melanjutkan replikasi, sel sering kali mengaktifkan polimerase khusus yang kurang teliti, disebut translesion synthesis polymerases. Enzim ini bisa melewati hambatan tersebut, tetapi dengan risiko tinggi memasangkan basa yang salah di seberang dimer.
- Ketiga, misalnya, di seberang dimer TT, polimerase yang tidak teliti mungkin memasangkan adenin (A) daripada adenin yang seharusnya berpasangan dengan setiap timin. Ini menghasilkan perubahan basa pada untai baru.
- Keempat, setelah replikasi selesai, sel anak akan mewarisi untai DNA baru yang sudah mengandung basa salah tersebut. Pada putaran replikasi berikutnya, basa salah ini akan diperlakukan sebagai templat yang benar, sehingga kesalahan diperkuat dan menjadi mutasi titik yang tetap (misalnya, situs asli TT di templat menjadi AA di turunannya).
Pertahanan Bawaan Sel Kulit Menghadapi Serangan Fotonik dari Matahari
Sel kulit kita bukanlah korban yang pasif. Mereka dilengkapi dengan sistem pengawasan dan tanggap darurat molekuler yang sangat canggih untuk menghadapi serangan UV. Begitu kerusakan DNA terdeteksi, alarm berbunyi di dalam nukleus, memicu kaskade pensinyalan yang bertujuan untuk menghentikan segala aktivitas seluler dan memprioritaskan perbaikan. Sistem ini adalah garis pertahanan pertama mencegah mutasi menjadi warisan permanen bagi sel-sel keturunan.
Kaskade Respons Seluler Pasca-Deteksi Kerusakan DNA
Deteksi awal dimulai oleh kompleks protein sensor yang terus-menerus memindai DNA. Protein seperti XPC dan DDB2 secara khusus dirancang untuk mengenali distorsi pada heliks DNA, seperti yang disebabkan oleh dimer pirimidin. Begitu mereka menemukan kerusakan, mereka menempel kuat di lokasi tersebut dan merekrut serangkaian protein lainnya. Salah satu peristiwa kunci berikutnya adalah fosforilasi (penambahan gugus fosfat) pada protein penjaga bernama p53.
Fosforilasi ini mengaktifkan p53, yang kemudian berfungsi sebagai “konduktor” respons darurat.
p53 yang aktif akan memicu dua jalur kritis secara paralel. Pertama, ia mengaktifkan gen yang menghambat siklus sel, khususnya di checkpoint G1/S. Ini dilakukan dengan meningkatkan produksi protein inhibitor seperti p21. Protein p21 akan mengikat dan menonaktifkan kompleks enzim yang mendorong sel masuk ke fase sintesis DNA. Hasilnya adalah jeda atau arrest siklus sel.
Pemberhentian ini memberikan waktu berharga bagi mesin perbaikan untuk bekerja sebelum replikasi dimulai. Kedua, p53 juga mengaktifkan gen-gen yang terlibat langsung dalam perbaikan DNA, seperti komponen sistem NER. Jika kerusakan terlalu parah dan tidak dapat diperbaiki, p53 akan memicu program apoptosis, atau kematian sel terprogram, untuk mengeliminasi sel yang berpotensi berbahaya secara elegan.
Komponen Kunci Sistem Perbaikan Eksisi Nukleotida (NER)
Sistem NER adalah regu darurat molekuler dengan spesialisasi yang terkoordinasi. Setiap anggota tim memiliki fungsi spesifik untuk memastikan kerusakan dipotong dan diganti dengan presisi.
- XPC-RAD23B: Tim pencari dan pengenal awal. Mereka secara konstan berpatroli, mendeteksi distorsi pada heliks DNA dan menandai lokasi kerusakan untuk tim perbaikan.
- TFIIH: Tim pembuka akses. Kompleks multi-protein ini menggunakan energi ATP untuk bertindak seperti motor molekuler, memutar DNA dan membuka heliks ganda di sekitar lokasi kerusakan, menciptakan “gelembung” untuk memberi ruang kerja.
- XPA & RPA: Manajer lapangan dan penstabil. XPA memverifikasi kerusakan dan membantu merekrut enzim pemotong. RPA menstabilkan untai DNA tunggal yang telah terbuka agar tidak terurai atau rusak.
- XPG & ERCC1-XPF Tim pemotong presisi. XPG memotong untai yang rusak di sisi 3′ dari kerusakan, sementara ERCC1-XPF memotong di sisi 5′. Potongan ini melepaskan segmen DNA sekitar 24-32 nukleotida yang mengandung dimer.
- DNA Polimerase δ/ε & PCNA: Tim rekonstruksi. Setelah segmen rusak dibuang, DNA polimerase yang akurat ini menggunakan untai komplemen yang sehat sebagai cetakan untuk mensintesis ulang nukleotida yang hilang.
- DNA Ligase: Tukang segel akhir. Enzim ini menyambungkan ujung nukleotida baru dengan ujung untai DNA yang lama, menutup celah dan menyelesaikan perbaikan.
Ilustrasi Presisi Kompleks Perbaikan
Bayangkan kompleks NER sebagai sebuah tim ahli bedah mikroskopis yang bekerja pada untaian DNA yang lebih tipis dari sehelai benang. Setelah protein sensor XPC menandai lokasi “tumor” (dimer), kompleks TFIIH mendekat. Dengan membakar molekul ATP sebagai bahan bakar, subunit protein TFIIH yang bernama XPB dan XPD mulai berputar, secara fisik memuntir DNA dan memisahkan kedua untainya di sekitar lokasi kerusakan, membentuk bukaan seperti gelembung selebar sekitar 20 nukleotida.
Di dalam gelembung ini, untai yang rusak menjadi terbuka. XPA kemudian masuk untuk memastikan posisi kerusakan tepat di tengah area kerja. RPA segera membungkus untai sehat di seberangnya untuk melindunginya. Dengan semuanya siap, dua enzim nuklease—XPG dan ERCC1-XPF—bergerak ke posisi yang tepat. Dengan gerakan terkoordinasi, seperti gunting bedah molekuler, mereka memotong untai yang rusak dengan jarak tepat beberapa nukleotida di hulu dan hilir dari dimer.
Segmen cacat itu kemudian terlepas. Ruang kosong yang ditinggalkan segera diisi oleh DNA polimerase yang menarik nukleotida baru dari pool dalam sel, mencocokkannya sempurna dengan untai templat yang sehat. Proses ini membutuhkan energi besar dari hidrolisis ATP untuk setiap langkah, menjamin akurasi di tingkat atom.
Contoh Mutasi pada Gen p53 yang Sering Lolos
Ironisnya, gen p53 yang menjadi sentral pertahanan ini sendiri adalah target utama mutasi yang diinduksi UV. Mutasi spesifik pada kodon 278 dari gen p53, yang mengubah basa sitosin menjadi timin, adalah tanda tangan klasik dari paparan sinar matahari. Mutasi ini sering terjadi karena sitosin dalam konteks sekuens tertentu (CpG) dapat termodifikasi dan menjadi lebih rentan membentuk dimer atau mengalami deaminasi setelah terpapar UV.
Ketika mutasi terjadi pada kedua alel gen p53 di sebuah sel, fungsinya sebagai penjaga genom hilang. Sel tersebut kehilangan kemampuan untuk menghentikan siklus sel saat ada kerusakan DNA dan juga kemampuan untuk memulai apoptosis. Akibatnya, sel dengan DNA rusak justru terus membelah, mengumpulkan lebih banyak mutasi. Inilah langkah kritis dalam transformasi sel normal menjadi sel prakanker, dan mengapa karsinoma sel skuamosa sering menunjukkan mutasi p53 yang spesifik akibat UV.
Dampak Terselubung Paparan UV Kronis Rendah terhadap Akumulasi Kesalahan Genetik
Banyak orang berpikir bahwa selama kulit tidak memerah atau terasa perih, paparan sinar matahari itu aman. Ini adalah pemahaman yang keliru. Paparan harian yang rendah, yang tidak sampai memicu sunburn atau bahkan disadari, sebenarnya memberikan “dosis kecil” kerusakan DNA secara terus-menerus. Seperti tetesan air yang melubangi batu, akumulasi kerusakan kecil ini selama puluhan tahun menciptakan beban mutasi yang signifikan di dalam sel-sel kulit, jauh sebelum kanker terlihat secara klinis.
Mekanisme di balik ini adalah bahwa bahkan pada intensitas rendah, foton UV tetap mampu menghasilkan dimer pirimidin, meski dalam jumlah yang lebih sedikit per waktu paparan. Yang menjadi masalah adalah kapasitas perbaikan sel kita tidak tak terbatas. Setiap hari, sel kulit harus memperbaiki kerusakan dari berbagai sumber, termasuk UV, radikal bebas dari metabolisme, dan lain-lain. Paparan UV kronis rendah secara konstan menambah beban kerja sistem perbaikan ini.
Seiring waktu, ada kemungkinan bahwa beberapa kerusakan lolos dari perbaikan, atau diperbaiki dengan kurang sempurna. Kesalahan kecil yang tertinggal ini, ketika sel membelah, akan dikunci menjadi mutasi permanen. Proses ini terjadi secara diam-diam, tanpa gejala inflamasi seperti sunburn, sehingga sering diabaikan.
| Jenis Sel Kulit | Sensitivitas Relatif terhadap UV | Waktu Bertahan Hidup / Siklus | Potensi Akumulasi Mutasi |
|---|---|---|---|
| Keratinosit Basal (Lapisan terdalam epidermis) | Tinggi (langsung menerima UV, sel punca/progenitor) | Bulanan (terus membelah untuk regenerasi) | Sangat Tinggi. Sebagai sel punca yang membelah terus, mutasi yang lolos akan diwariskan ke banyak sel anak dan menetap lama di jaringan. |
| Melanosit (Penghasil melanin) | Sedang, tetapi memiliki perlindungan melanin internal. | Tahun hingga permanen (jarang membelah) | Kritis. Mutasi pada gen pengendali siklus sel (seperti pada melanoma) bisa sangat berbahaya karena sel ini berumur panjang. |
| Sel Langerhans (Sel imun) | Sangat Tinggi (sangat rentan terhadap apoptosis oleh UV) | Minggu hingga bulan | Rendah secara langsung, tetapi tinggi secara tidak langsung. Kematian mereka melemahkan imunosurveillance, memungkinkan sel lain yang bermutasi berkembang. |
Dalam fotokarsinogenesis, mutasi akibat UV dapat dikategorikan menjadi “driver” dan “passenger”. Mutasi driver adalah perubahan pada gen-gen penting yang mengendalikan pertumbuhan, pembelahan, dan kematian sel (seperti p53, RAS, atau gen penekan tumor lainnya). Mutasi ini memberikan keuntungan selektif kepada sel, membuatnya tumbuh lebih cepat dan menghindari kematian. Sementara mutasi passenger adalah perubahan pada gen lain yang tidak memberikan keuntungan langsung, hanya ikut terbawa karena kerusakan DNA di sekitarnya. UV bertindak sebagai inisiator yang diam-diam dengan secara acak menciptakan puluhan hingga ratusan mutasi, termasuk beberapa yang berpotensi menjadi driver. Sel yang kebetulan mendapatkan mutasi driver itulah yang kemudian dapat berkembang menjadi klonal dan memulai proses kanker.
Peran Lingkungan Mikro dan Penuaan, Paparan sinar UV tinggi pada sel dapat mengakibatkan mutasi
Akumulasi kesalahan genetik ini dipercepat oleh dua faktor utama: lingkungan mikro seluler dan penuaan biologis. Seiring bertambahnya usia, efisiensi semua sistem perbaikan DNA, termasuk NER, secara alami menurun. Aktivitas enzim-enzim kunci berkurang, dan akumulasi kerusakan oksidatif internal juga mengganggu fungsi seluler. Ini berarti sel kulit orang yang lebih tua kurang mampu memperbaiki kerusakan UV yang terjadi hari ini dibandingkan sel kulit orang muda.
Selain itu, lingkungan mikro kulit yang menua juga berubah. Produksi kolagen menurun, sirkulasi mungkin tidak sebaik dulu, dan komunikasi antarsel bisa kurang optimal. Perubahan ini menciptakan lingkungan yang lebih permisif bagi sel-sel yang telah mengakumulasi mutasi untuk bertahan dan berkembang, karena sistem pengawasan jaringan dan imun juga tidak seketat dulu. Kombinasi dari paparan UV kronis rendah, penurunan kapasitas perbaikan, dan lingkungan mikro yang mendukung inilah yang menjelaskan mengapa sebagian besar kanker kulit muncul pada usia lanjut.
Interaksi Silang antara Radiasi UV dan Agen Kimiawi dalam Memperbesar Cacat Genetika
 ~ Info Untuk Kita")
Source: ac.id
Risiko mutasi dari sinar matahari tidak berdiri sendiri. Dalam kehidupan sehari-hari, kulit kita juga terpapar berbagai senyawa kimia dari lingkungan, produk perawatan, atau bahkan makanan. Beberapa senyawa ini, ketika bertemu dengan radiasi UV, dapat bertindak sebagai mitra kejahatan yang memperparah kerusakan DNA. Interaksi sinergis ini seringkali diabaikan, padahal dampaknya bisa jauh lebih besar daripada efek masing-masing faktor secara terpisah. Kombinasi ini tidak hanya menambah jumlah kerusakan, tetapi juga dapat menciptakan jenis kerusakan yang lebih rumit dan sulit diperbaiki oleh sel.
Mekanisme sinergi ini beragam. Beberapa senyawa kimia berperan sebagai photosensitizer. Contoh klasik adalah psoralen, senyawa yang ditemukan dalam beberapa tanaman seperti jeruk dan seledri, dan digunakan dalam terapi PUVA untuk psoriasis. Psoralen dapat masuk ke dalam sel dan menyelip di antara basa DNA. Ketika terpapar UV-A, psoralen yang tereksitasi akan bereaksi langsung dengan basa pirimidin, membentuk adduct (ikatan kimia) antar-untai DNA.
Adduct ini mengikat kedua untai DNA secara bersamaan, membuatnya sangat sulit diperbaiki karena menghalangi proses pemisahan untai yang diperlukan untuk perbaikan. Agen lain, seperti arsenik yang mungkin terkandung dalam air tanah, tidak langsung merusak DNA bersama UV, tetapi bekerja dengan cara mengganggu sistem perbaikan DNA itu sendiri. Arsenik dapat menghambat enzim-enzim kunci dalam jalur perbaikan, mengurangi kapasitas sel untuk membersihkan dimer pirimidin yang dihasilkan UV, sehingga mutasi lebih mudah terkunci.
Tahapan Molekuler Peningkatan Sensitivitas Sel
Interaksi antara agen kimia dan UV dalam memperbesar cacat genetik terjadi melalui beberapa tahapan molekuler yang saling terkait:
- Pembentukan Adduct DNA yang Kompleks: Photosensitizer seperti psoralen membentuk monoadduct atau crosslink (ikatan silang) antar-untai DNA setelah diaktifkan oleh UV-A. Struktur ini lebih besar dan lebih distorsif daripada dimer timin biasa.
- Penghambatan Enzim Perbaikan: Logam berat seperti arsenik atau kadmium dapat berikatan dengan gugus sulfhidril (-SH) pada protein, mengganggu struktur dan fungsi enzim-enzim NER seperti XPA atau protein yang terlibat dalam pensinyalan kerusakan DNA.
- Peningkatan Stres Oksidatif: Beberapa senyawa kimia, ketika terkena UV, dapat memicu produksi radikal bebas dalam jumlah besar di dalam sel, menyebabkan kerusakan DNA oksidatif tambahan yang membanjiri sistem perbaikan eksisi basa.
- Gangguan pada Checkpoint Siklus Sel: Agen kimia tertentu dapat mengganggu pensinyalan protein seperti p53 atau ATM/ATR, yang bertugas menghentikan siklus sel saat ada kerusakan. Gangguan ini membuat sel terus membelah meski DNA-nya penuh dengan lesi yang belum diperbaiki.
Ilustrasi Pembanjiran Kapasitas Pertahanan Sel
Bayangkan sistem pertahanan DNA sel sebagai sebuah tim pemadam kebakaran yang sangat terlatih di sebuah kota (sel). Paparan UV saja seperti serangkaian kebakaran kecil di beberapa rumah (dimer). Tim pemadam dapat menangani mereka satu per satu. Namun, paparan simultan dari agen kimia photosensitizer adalah seperti seseorang yang menyebarkan bahan bakar dan petasan di seluruh kota tepat sebelum kebakaran kecil itu muncul.
Saat UV (pemicu api) datang, yang terjadi bukanlah kebakaran kecil, tetapi ledakan dan kebakaran hebat di mana-mana (adduct kompleks dan crosslink). Pada saat yang sama, arsenik bertindak seperti seseorang yang merusak selang air, mematikan pompa, dan mengacaukan komunikasi radio tim pemadam. Akibatnya, sistem yang biasanya efisien menjadi kewalahan total. Kebakaran yang tidak tertangani (kerusakan DNA) merajalela, merusak struktur vital kota (gen penting), dan meninggalkan kerusakan permanen yang jauh lebih parah daripada jika hanya ada satu ancaman saja.
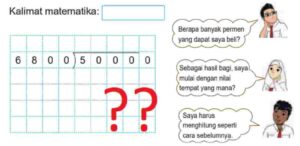
Paparan sinar UV tinggi pada sel dapat mengakibatkan mutasi, sebuah fenomena biologis yang serius. Proses analisis seperti ini mirip dengan ketika kita mengevaluasi data, misalnya saat membaca Perbandingan Kinerja Kelas A dan B pada Tes. Keduanya memerlukan ketelitian untuk melihat perbedaan dan dampaknya. Nah, kembali ke topik, mutasi DNA akibat UV ini bisa berakibat panjang, mengubah fungsi sel secara fundamental dan berpotensi memicu masalah kesehatan yang serius.
Inilah yang membuka jalan bagi mutasi kompleks dan berbahaya, seperti delesi besar atau translokasi kromosom, yang lebih mungkin mengaktifkan onkogen atau menonaktifkan gen penekan tumor.
| Agen Kimia | Sumber Paparan Umum | Mekanisme Interaksi dengan UV | Jenis Mutasi yang Dihasilkan |
|---|---|---|---|
| Psoralen (Furocoumarins) | Tanaman (jeruk, seledri, parsnip), terapi PUVA. | Photosensitizer; membentuk adduct dan crosslink DNA setelah diaktifkan UV-A. | Mutasi titik, delesi, dan yang paling khas: mutasi pada gen p53 spesifik untuk crosslink. |
| Arsenik | Air tanah yang tercemar, industri tertentu. | Mengganggu pensinyalan dan perbaikan DNA; menghambat enzim NER; sinergi dengan UV sebagai co-carcinogen. | Meningkatkan frekuensi semua mutasi yang diinduksi UV; terkait dengan karsinoma sel skuamosa dan melanoma. |
| Polycyclic Aromatic Hydrocarbons (PAHs) | Asap rokok, polusi udara, makanan yang dibakar. | Produk metaboliknya (diol epoxide) membentuk adduct DNA; UV meningkatkan stres oksidatif dan dapat mengganggu perbaikan adduct ini. | Mutasi transisi G→T yang khas, sering pada gen seperti p53 dan RAS. |
| Beberapa Obat Topikal (mis., tertentu antibiotik, agen kemoterapi) | Pengobatan kulit. | Dapat bertindak sebagai photosensitizer lokal, menyebabkan fototoksisitas dan kerusakan DNA yang diperantarai radikal bebas saat terpapar sinar matahari. | Kerusakan DNA nonspesifik, berpotensi memicu mutasi di lokasi aplikasi. |
Evolusi Adaptasi Molekuler pada Organisme yang Hidup di Lingkungan UV Ekstrem
Sementara sel manusia berjuang melawan dampak UV, beberapa organisme justru berkembang dan bahkan berkembang biak di lingkungan dengan radiasi UV yang mematikan. Tempat-tempat seperti puncak gunung tinggi, gurun yang terik, atau daerah yang terkontaminasi radiasi, menjadi rumah bagi ekstremofil seperti bakteri Deinococcus radiodurans. Organisme ini tidak kebal terhadap kerusakan DNA; sebaliknya, DNA mereka dihancurkan berkeping-keping oleh paparan intens. Rahasia mereka bukanlah pencegahan, tetapi kemampuan luar biasa untuk menyatukan kembali genom mereka yang hancur dengan cepat dan sempurna.
Mempelajari mekanisme pertahanan alami mereka membuka jendela baru untuk memahami ketahanan genetik dan potensi aplikasi biomedis.
Deinococcus radiodurans adalah contoh paling terkenal. Bakteri ini dapat bertahan dari dosis radiasi gamma atau UV ribuan kali lebih tinggi daripada yang dapat dibunuh manusia. Setelah paparan yang begitu ekstrem, kromosomnya terfragmentasi menjadi ratusan potongan. Dalam hitungan jam, bakteri ini mampu merakit ulang genomnya secara utuh dan melanjutkan pembelahan sel. Kunci utamanya terletak pada kombinasi strategi yang meliputi struktur sel yang protektif, sistem perbaikan DNA yang sangat efisien, dan lingkungan sitoplasma yang dirancang untuk meminimalkan kerusakan sekunder.
Mereka telah mengembangkan toolkit molekuler super melalui miliaran tahun evolusi di kondisi marginal.
Mekanisme Ketahanan Ekstremofil
Toolkit molekuler organisme tahan UV ini terdiri dari beberapa komponen yang bekerja sinergis:
- Struktur Sel dan Pengemasan DNA yang Unik: D. radiodurans memiliki dinding sel yang tebal dan struktur sitoplasma yang membuat kromosomnya tetap tertata rapat, mungkin dalam bentuk toroid (cincin). Pengemasan ini mencegah potongan DNA yang terfragmentasi tersebar terlalu jauh, memudahkan proses reassembly. Beberapa organisme juga memiliki pigmen seperti scytonemin (pada cyanobacteria) yang bertindak sebagai tabir surya alami, menyerap energi UV sebelum mencapai DNA.
- Sistem Perbaikan Rekombinasi Homolog yang Super Efisien: Ini adalah bintang utama. Organisme ini memiliki banyak salinan genomnya (polisomi) selama fase pertumbuhan tertentu. Ketika satu salinan hancur, mereka menggunakan salinan lainnya sebagai cetakan yang utuh untuk memperbaiki kerusakan. Enzim-enzim perbaikannya, seperti RecA, bekerja dengan kecepatan dan ketepatan yang luar biasa dalam menyambung potongan-potongan DNA berdasarkan homologi sekuens.
- Pembersihan Radikal Bebas yang Agresif Sitoplasma mereka dipenuhi dengan konsentrasi tinggi antioksidan seperti mangan kompleks dan enzim katalase/superoxide dismutase. Ini dengan cepat menetralkan spesies oksigen reaktif yang dihasilkan oleh radiasi, mencegah kerusakan oksidatif sekunder pada protein dan DNA yang sedang diperbaiki.
- Protein Protektif dan Perbaikan Multi-Jalur: Mereka mengekspresikan protein khusus yang melindungi DNA dari denaturasi dan mengikat potongan DNA. Selain itu, mereka tidak hanya mengandalkan satu jalur perbaikan seperti NER, tetapi memiliki dan mengkoordinasikan multiple jalur (excision repair, recombination, end-joining) secara simultan.
Adaptasi dari ekstremofil seperti Deinococcus radiodurans memberikan pelajaran berharga untuk penelitian biomedis manusia. Pertama, ini membuktikan bahwa perbaikan DNA masif dengan akurasi tinggi secara biologis dimungkinkan. Para ilmuwan sedang mempelajari enzim-enzim perbaikannya untuk potensi aplikasi dalam bioteknologi, seperti pengeditan gen yang lebih aman. Kedua, strategi mereka dalam mengelola stres oksidatif (melalui kompleks mangan) menginspirasi penelitian tentang antioksidan baru yang lebih efektif untuk melindungi sel kulit.
Ketiga, memahami bagaimana mereka menjaga integritas kromosom meski terfragmentasi dapat memberikan petunjuk untuk mengembangkan terapi yang meningkatkan ketahanan sel sehat selama radioterapi kanker atau untuk melindungi astronaut dari radiasi kosmik.
| Aspek Pertahanan | Sel Kulit Manusia (Normal) | Ekstremofil (contoh: Deinococcus radiodurans) |
|---|---|---|
| Strategi Inti | Deteksi dini & perbaikan cepat sebelum replikasi; apoptosis jika gagal. | Toleransi kerusakan masif & reassembly genom pasca-kehancuran. |
| Perbaikan DNA Andalan | NER untuk lesi besar seperti dimer; BER untuk kerusakan oksidatif. | Rekombinasi Homolog yang sangat cepat dan akurat, didukung oleh genom ganda. |
| Perlindungan Fisik | Stratum korneum (lapisan kulit mati), melanin. | Dinding sel tebal, pengemasan DNA toroidal, pigmen UV-absorbing (scytonemin). |
| Manajemen Stres Oksidatif | Sistem antioksidan standar (glutathione, SOD, katalase). | Konsentrasi sangat tinggi dari kompleks mangan antioksidan dan enzim pembersih radikal. |
| Respons terhadap Kerusakan Parah | Penghentian siklus sel dan apoptosis. | Penghentian sementara diikuti mobilisasi masif semua sistem perbaikan untuk reassembly. |
Ulasan Penutup
Jadi, setelah menyelami dunia di balik sel yang terpapar UV, satu hal menjadi jelas: sinar matahari membawa cerita yang lebih kompleks daripada sekadar kulit terbakar atau warna tan yang ideal. Ia adalah pemain kuat dalam narasi kesehatan seluler jangka panjang kita. Mulai dari mekanisme perbaikan yang heroik hingga akumulasi kesalahan diam-diam yang mengintai, pemahaman ini bukan untuk membuat kita takut pada matahari, tetapi untuk menghargai kompleksitas pertahanan tubuh dan pentingnya perlindungan yang cerdas.
Alam bahkan mengajarkan kita melalui ekstremofil bahwa ketahanan adalah mungkin, menawarkan secercah harapan untuk inovasi masa depan. Intinya, menghormati matahari berarti memahami kekuatannya yang halus namun mendalam dalam menulis ulang cerita di tingkat seluler.
Pertanyaan yang Sering Muncul: Paparan Sinar UV Tinggi Pada Sel Dapat Mengakibatkan Mutasi
Apakah semua jenis sinar UV dari matahari berbahaya dan menyebabkan mutasi?
Tidak semua sama berbahayanya. UV-C paling energik namun biasanya disaring oleh atmosfer. UV-B adalah penyebab utama pembentukan dimer pirimidin dan kerusakan DNA langsung. Sementara UV-A, meski kurang energik, dapat menembus lebih dalam, menghasilkan radikal bebas yang merusak DNA secara tidak langsung dan berperan dalam penuaan kulit serta kanker.
Apakah tabir surya (sunscreen) benar-benar bisa mencegah kerusakan DNA dan mutasi ini?
Ya, secara signifikan. Tabir surya dengan SPF dan broad-spectrum (melindungi dari UV-A & UV-B) bertindak sebagai perisai fisik atau kimiawi yang mengurangi jumlah foton UV yang mencapai sel kulit. Dengan mengurangi paparan, beban kerusakan DNA pada mekanisme perbaikan sel menjadi lebih ringan, sehingga menurunkan risiko mutasi yang tidak terperbaiki.
Jika sel punya mekanisme perbaikan yang canggih, mengapa mutasi tetap bisa terjadi?
Mekanisme perbaikan, seperti NER, sangat efisien tapi tidak sempurna. Kapasitasnya bisa kewalahan oleh paparan UV tinggi atau kronis. Selain itu, efisiensinya menurun seiring usia. Terkadang, perbaikan juga bisa membuat kesalahan, atau kerusakan terjadi di area DNA yang “terlupakan” oleh sistem pengawasan sel. Mutasi kecil yang lolos ini dapat terakumulasi seumur hidup.
Apakah mutasi akibat UV selalu berujung pada kanker kulit?
Tidak selalu. Tubuh memiliki banyak lapisan pertahanan. Sebagian besar mutasi bersifat “passenger” yang tidak memberikan keuntungan pertumbuhan bagi sel. Namun, jika mutasi “driver” yang kritis—misalnya pada gen penekan tumor seperti p53—terjadi dan lolos dari semua pengawasan, sel tersebut berpotensi memulai perjalanan menuju transformasi ganas. Risikonya meningkat dengan jumlah dan frekuensi paparan.
Bisakah kerusakan DNA akibat UV yang sudah terjadi diperbaiki setelah paparan selesai?
Ya, dalam batas tertentu. Sistem perbaikan DNA seperti NER bekerja segera setelah kerusakan terdeteksi dan terus aktif setelah paparan. Namun, prosesnya membutuhkan waktu. Jika paparan berikutnya terjadi sebelum perbaikan tuntas, kerusakan baru akan menumpuk di atas yang lama. Inilah mengapa perlindungan berkelanjutan dan memberi waktu pemulihan pada kulit sangat penting.